近日,中国海洋大学医药学院、海洋药物教育部重点实验室、青岛海洋科技中心海洋药物与生物制品功能实验室王杨教授课题组和卢玲教授课题组,在卤代腙的可控环化反应开发和新型吲哚啉类HIF抑制剂创制研究方面取得重要进展,相关研究论文以“Switchable Reactivity of Hydrazonoyl Halides: Enabling the Discovery of Potent Hypoxia-Inducible Factor Inhibitors(卤代腙的可控反应性:强效缺氧诱导因子抑制剂的发现)”为题发表于国际知名学术期刊JACS Au。
卤代腙类化合物具有独特的C=N−N骨架,是一类多功能合成砌块构建多种含氮杂环。相较于研究较为成熟的[3+2]和[3+3]环加成反应,卤代腙参与的[3+4]环加成反应研究尚少。更重要的是,尚未有将其作为C1合成子用于环加成反应的报道。现有研究中,卤代腙的反应模式始终局限于甲亚胺碳表现为亲电性。若能实现卤代腙极性反转,将改变其固有反应路径,为具有新型复杂骨架的含氮杂环化合物的合成提供新途径,为获取具有药理活性的新化合物奠定物质基础。因此,调控卤代腙的反应性既是合成化学领域的重大挑战,也具有重要研究价值。针对该科学难题,王杨课题组发展了取代基调控的卤代腙与2-氨基查尔酮发散环加成新策略,通过2-氨基查尔酮底物氨基取代基变化精准切换反应路径:N-磺酰基取代底物发生形式[1+4]环加成,高效构建吲哚啉母核;游离NH2底物则经由形式[3+4]环加成,选择性合成苯并三氮杂䓬衍生物,首次实现卤代腙亚胺碳极性反转、作为C1合成子参与环加成,突破了该类化合物固有的反应模式局限。
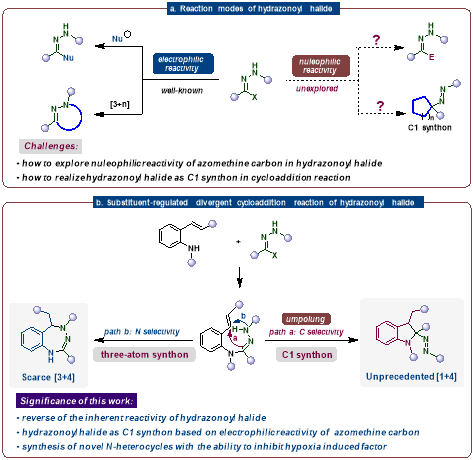
图1 卤代腙的反应模式和取代基调控的卤代腙多样性环化及产物的生物活性研究
当使用N-磺酰取代的2-氨基查尔酮时,卤代腙发生极性反转,作为单原子合成子与之发生[1+4]环化反应,生成一类全新的吲哚啉产物。对具有不同取代基的2-氨基查尔酮底物和卤代腙底物进行考察后发现,其适用性均较好,能以优秀的非对映选择性(dr > 19:1)得到目标产物。其中,2-氨基查尔酮的中心苯环和不饱和酮片段末端芳环上连接吸电子或给电子基时对反应无明显影响,甚至当不饱和酮末端为芳杂环或脂肪取代基时,反应仍能顺利进行。氨基上不同种类的磺酰基取代均不会影响反应的进行。对卤代腙底物进行考察后发现,苯环上不同位置不同电性取代的卤代腙底物均表现出了良好的适用性。
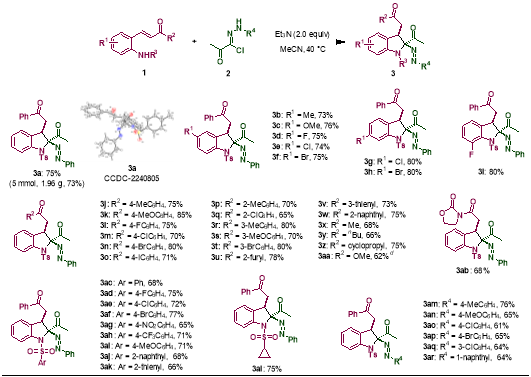
图2 [1+4]环化反应的底物拓展
另一方面,当以无取代的2-氨基查尔酮底物进行反应时,卤代腙底物作为三原子合成子与之发生[3+4]环化反应。在2-氨基查尔酮的中心芳环上引入一系列取代基均能良好耐受,在其Michael受体片段中,末端的对位、间位和邻位取代的苯基以及萘基和杂芳环等各类官能团均表现出良好的兼容性。当连接在Michael受体上的芳香基团被烷基取代时,反应同样能顺利进行。最后,对卤代腙底物的苯环上引入了多种取代基,均能高效地实现[3+4]环加成产物的成功构建。
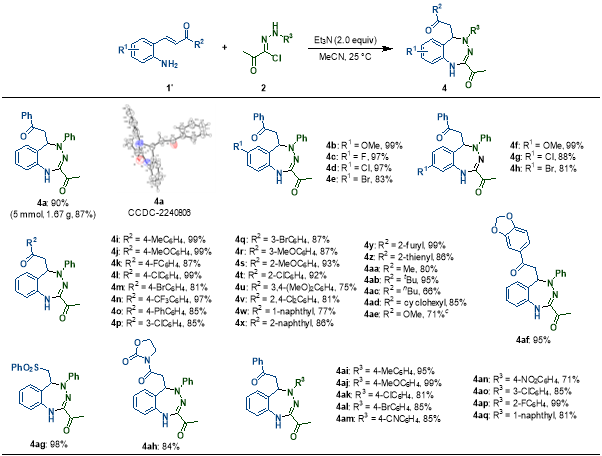
图3 [3+4]环化反应的底物拓展
作者结合实验机理研究与密度泛函理论(DFT)计算,系统阐明了取代基调控反应选择性的本质:环加成步骤为反应决速步与选择性决定步;对甲苯磺酰基与乙酰基在七元环过渡态中的空间位阻效应,导致共轭作用消失、能垒升高,最终实现反应路径反转。该理论计算结果与实验现象一致,为反应性调控提供了坚实理论支撑。
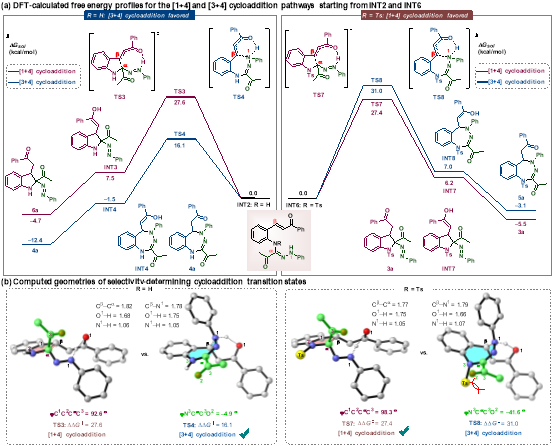
图4 反应机理验证
HIF是调控肿瘤低氧微环境的核心转录因子,其异常高表达与肿瘤增殖、侵袭、转移密切相关,是抗肿瘤药物研发的重要靶点。依托该极性反转策略合成的新型吲哚啉化合物,展现出优异的低氧诱导因子(HIF)抑制活性。体外活性筛选显示,吲哚啉化合物3ac对HIF转录活性的IC50低至3.82 μM,且在HCT116、HEK293T、Hela等多种细胞中细胞毒性极低(IC50>100 μM),实现了高效抑制与低毒性的理想平衡。
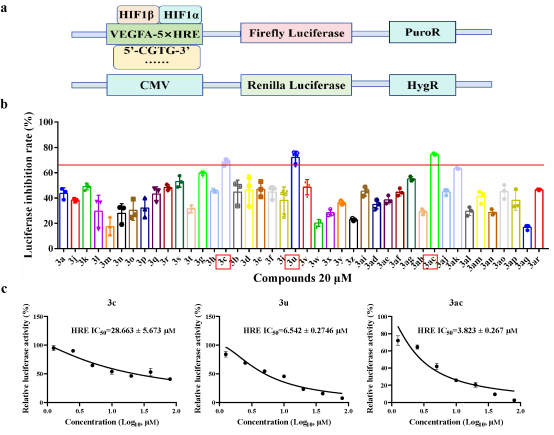
图5 化合物3ac在体外实验中展现出了缺氧诱导因子抑制活性
机制研究证实,化合物3ac可显著降低HIF1α、HIF2α蛋白水平,并抑制其下游靶基因CA9、REDD1、HK2等的mRNA表达,直接作用于HIF1α转录激活结构域(TAD),阻断其转录功能。小鼠体内抗肿瘤实验表明,3ac可显著抑制结肠癌CT26移植瘤生长,且对小鼠体重无明显影响,具备良好的体内安全性与成药潜力。
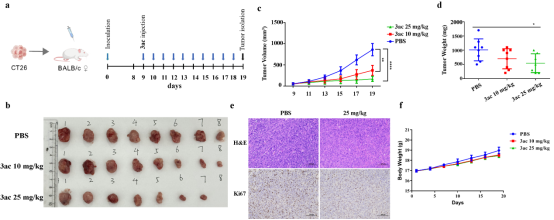
图6 化合物3ac在体内实验中能够抑制肿瘤的生长
本文的第一完成单位是中国海洋大学,医药学院王杨教授、卢玲教授和浙江工业大学的杨云芳教授为共同通讯作者。文章的第一作者是中国海洋大学的田晓晨,浙江工业大学的吴洪丽和中国海洋大学的石耀宇、崔晓楠、庞若尘、张博奇为文章共同作者。该工作得到了泰山学者青年专家、国家自然科学基金等项目的支持。
【关闭】





