1-磷酸鞘氨醇受体2 (S1PR2)是G蛋白偶联受体家族成员之一,由7段跨细胞质膜的α螺旋结构组成,广泛分布于全身各处组织中。当特定配体与之结合后,会引起G蛋白(Gαs,Gαq和Gα12/13)的活化,产生第二信使和效应器蛋白将所接受的胞外信号向下游传递,进而在多种生理过程和疾病进展中发挥关键作用。并且在近几年中,S1PR2得到了研究者的广泛关注,其激活后参与癌症、炎症、纤维化疾病、多发性硬化和5-FU癌症耐药等多种类型疾病的发展。另外,S1PR2对肿瘤生长和进展的影响具有特异性,激活或抑制S1PR2产生的效果取决于所研究的疾病机制。因此,调节S1PR2的激活被认为是治疗多种疾病的一种很有前景的策略。近日,中国海洋大学医药学院、海洋药物教育部重点实验室、青岛海洋生物医药研究院李晓杨课题组在药学顶期《Medicinal Research Reviews》(IF = 13.3)上发表题为“An Overview of Sphingosine-1-phosphate Receptor 2: Structure, Biological Function, and Small-molecule Modulators”的综述,详述了S1PR2的结构、生理学功能及其在人类疾病中的病理作用,并系统论述了近年来团队与合作研究人员对S1PR2调节剂的发现方法、设计策略、开发过程和生物医学应用。(文章链接:http://doi.org/10.1002/med.22044)。
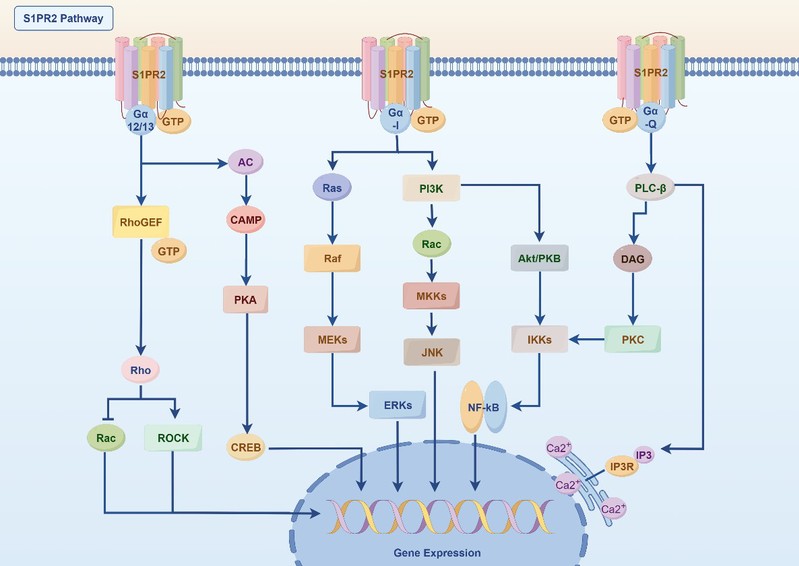
图1. S1PR2的关键信号网络
迄今为止,已经开发了大量针对S1PR2的拮抗剂和少量激动剂,它们被用作体内成像的化学工具分子或用于治疗各种疾病的先导化合物,有4种S1PR2拮抗剂在临床前实验中显示出较好的效果,这突出了它们潜在的治疗应用。在此重点介绍了S1PR2调节剂的产生与发展。其中,S1PR2激活剂的设计包括基于S1P和胆汁酸等正构配体形成的激活剂和通过高通量筛选所得的变构调节剂。S1PR2拮抗剂则主要是围绕经典的特异性S1PR2选择性拮抗剂JTE-013、高通量筛选和基于结构的设计策略等方式获得的先导化合物进行结构修饰和改造而形成。此外,我们概述了该领域的主要挑战和未来方向。首先是由于之前对S1PR2结构认知不清晰,缺乏基于结构的药物设计,所以大多数报道的S1PR2调节剂来源于内源性配体或HTS,其次是传统的药物化学优化。但该障碍已随着S1P-S1PR2复合物被阐明而解决,这有望极大地促进S1PR2激动剂和拮抗剂的发现。另一个挑战是对S1PR2在多种疾病中的作用机制的了解有限,这阻碍了对调节剂的生物学评价,所以对S1PR2机制的研究显得格外重要。随着S1PR2激动剂和拮抗剂根据不同策略的不断设计,对S1PR2功能的更好理解将会出现,这可能会加速一些S1PR2调节剂进入疾病治疗或诊断的临床研究。
中国海洋大学为第一通讯单位,李晓杨副教授为通讯作者,医药学院硕士研究生郝婉婷同学为第一作者,医药学院博士研究生罗栋栋(已毕业)为该综述共同第一和共同通讯作者,江余祺副教授和万升标教授为合作作者。该研究由国家自然科学基金、中央高校-中国海洋大学基本科研业务费专项资金、泰山学者奖励计划和山东省优秀青年科学基金等项目资助。
【关闭】





