近日,由中国海洋大学医药学院邵长伦教授领衔,联合多位合作者,在抗炎天然产物全合成方面取得新进展。相关研究成果以“Highly efficient and scalable total syntheses and stereochemical assignment of potent anti-inflammatory pesimquinolones I and J”(《抗炎pesimquinolones I 与J的高效、可拓展全合成及立体构型的确定》)为题,作为封面文章发表在Organic Chemistry Frontiers(《有机化学前沿》,图1)上。

图1. 工作以封面文章发表在Organic Chemistry Frontiers
活性天然产物作为药物先导化合物一直是药物研发的重要源泉。自首个3,4-二氧代-5-羟基-4-芳基-2(1H)-喹啉酮生物碱NTC-47被分离报道以来,该类新颖天然产物家族成员被不断从陆地和海洋真菌中发现。因其独特的结构和多样的生物活性引起了有机合成化学家和药物化学家的极大兴趣。邵长伦教授团队在前期3,4-二氧代-5-羟基-4-芳基-2(1H)-喹啉酮生物碱发现(J. Nat. Prod., 2014,77, 2720; Mar. Biotechnol., 2015, 17, 408; US Patent US10639303, 2020)与化学合成(Commun. Chem., 2022, 5, 35; Eur. J Med. Chem., 2023, 250, 115183;Tetrahedron, 2023,133299)基础上,报道了抗炎天然产物pesimquinolones I和J的高效、可扩展全合成及结构确定。
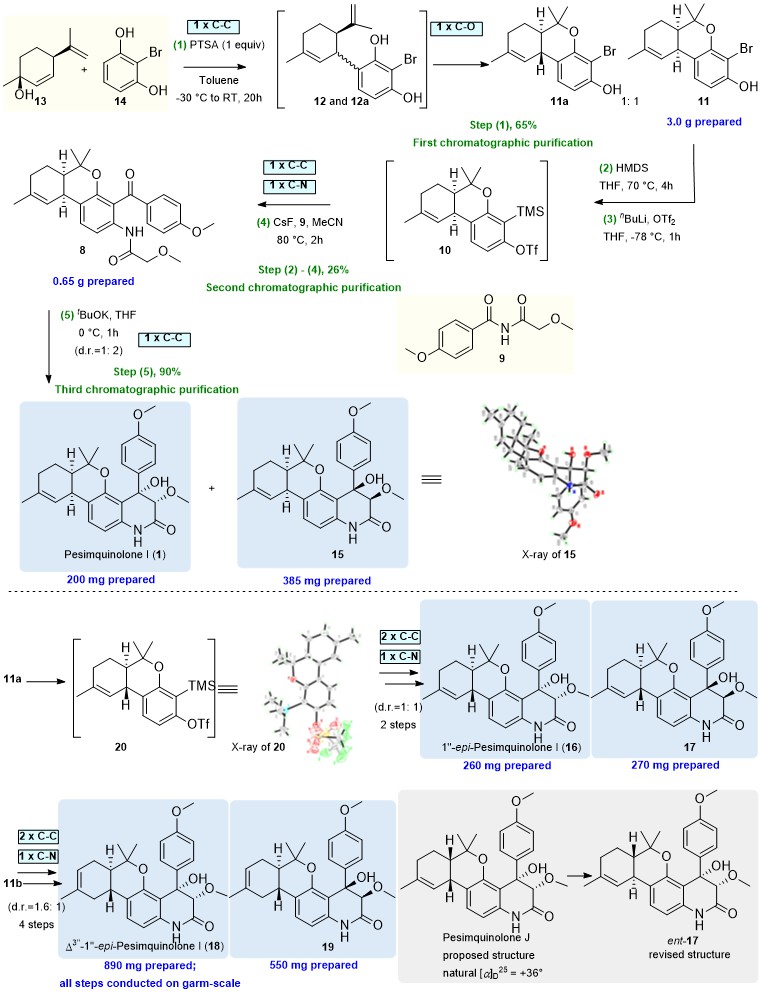
图2. Pesimquinolone I的全合成路线图
Pesimquinolone I的全合成路线图2所示。从合成关键中间体11开始,由商业可得的(1S,4R)-1-甲基-4-(1-甲基乙烯基)-2-环己烯-1-醇(13)和2-溴间苯二酚(14)为起始原料,通过条件优化筛选,最终以PTSA为酸,甲苯为溶剂,-30℃到室温的条件下,实现了顺式关键中间体11的大量制备(第一次柱层析分离)。在大量制备中间体11后,经过逆Brook重排反应顺利得到中间体10。接下来的挑战是芳炔插入反应来区域选择性一步实现两个化学键和喹啉酮母核的构建。在CsF存在下,中间体10与二酰亚胺9进行区域选择性的芳炔插入反应,以三步26%的收率成功构筑了二苯甲酮8(第二次柱层析分离)。随后使用分子内Aldol反应,在叔丁醇钾的碱性条件下,以非对映选择性、90%的收率合成了天然产物pesimquinolone I(1)及其非对映异构体15(第三次柱层析分离)。最终,以5步化学反应、3次柱层析分离、2.8%的总收率高效完成了天然产物pesimquinolones I的首次全合成。
巧合的是,化合物15是另外一个天然产物pesimquinolone J的对映异构体。然而,通过仔细对比相关数据后发现,它们的核磁数据有明显不同。推测是之前报道的天然产物C-1’’或C-6’’位的绝对构型有误。接下来又开展了其他立体异构体的合成工作,纠正并确定了pesimquinolone J的绝对构型。特别有意义的是,发现天然产物类似物18具有显著的抑制NO的作用,其IC50值为0.44 μM,远优于阳性药吲哚美辛(IC50 = 50.00 μM)。此外,该化合物还显示强的抑制促炎细胞因子TNF-α (p < 0.0001)和IL-6 (p < 0.0001) 的产生。以上研究表明,18具备开发成新型抗炎药物的潜力。
中国海洋大学为第一通讯单位,邵长伦教授为通讯作者,医药学院博士生郭凤伟为第一作者,王长云教授、顾玉诚博士、陈光英教授、魏美燕副教授、博士生周天意和硕士生曲勇等为共同作者。相关工作得到国家重点研发计划项目、国家自然科学基金、中央高校基本科研业务费和先正达博士生奖学金等项目的资助。
文章链接:https://pubs.rsc.org/en/content/articlepdf/2023/qo/d2qo01788a
https://mp.weixin.qq.com/s/BeHOBmC7Wcrtg8jQCovAuw
【关闭】





