来源于海洋生物资源的天然产物具有结构新颖、种类多样和靶向性强的特点,并在抗肿瘤、抗病毒、镇痛和抗菌等方面发挥着重要作用。其中,海洋多肽作为海洋天然产物的重要组成部分,因兼具抗体“靶向性”与小分子“易透过胞膜、易进行化学修饰”等的特点,受到了研究者们的广泛关注。特别是自固相合成技术兴起以来,多肽合成技术和多肽药物研发开始飞速发展。近日,中国海洋大学医药学院、海洋药物教育部重点实验室江涛教授和于日磊教授团队在多肽药物研发方面取得重要进展,于2022年9月26日在国际顶尖药学期刊Journal of Medicinal Chemistry在线发表关于芋螺毒素研究的重要成果(DOI: 10.1021/acs.jmedchem.2c00494),并被当选为当期杂志封面(图1)。
图1:芋螺毒素Mr1.1[S4Dap]具有开发为新型镇痛药物的潜力(文章封面)。
芋螺毒素(CTxs)是由海洋软体动物芋螺的毒液管和毒囊内壁的毒腺所分泌的一系列富含二硫键的生物活性肽,其能够特异性地靶向细胞膜上的不同离子通道受而具有重要的药用潜力。α9α10 乙酰胆碱受体(α9α10 nAChR)是新型镇痛药物开发的重要靶点,然而作用该受体的芋螺毒素往往具有物种选择性,对人源受体活性远远低于鼠源受体。实验室长期致力于发现不具物种选择性的新型芋螺毒素。研究人员根据前期研究发现,虽然已报道的α-芋螺毒素(α-CTxs)Vc1.1和Mr1.1分别是α9α10 乙酰胆碱受体和α7 nAChR的特异性抑制剂,但是其氨基酸序列十分相似,并且两者二硫键的连接方式是一致的,同时考虑到α7和α9亚基高度的序列同源性,一般对α9α10 nAChR有抑制作用的毒素对α7 nAChR也有一定的效果。因此研究人员猜测Mr1.1是否也能通过作用于α9α10 nAChR发挥抑制作用,从而在镇痛药开发方面打下一定的基础。接着,研究人员对Mr1.1进行了化学合成和电生理活性评价。结果表明:与其它nAChRs相比,Mr1.1对α9α10 nAChR具有更强的抑制效果,并强于Vc1.1(因物种选择性失败于临床2期研究)的活性,具有很好的镇痛药研发前景。
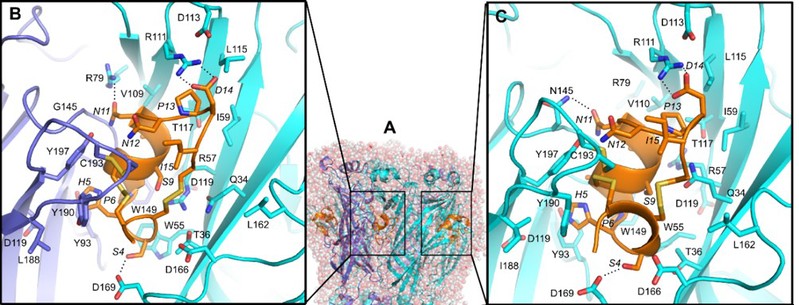
图2:芋螺毒素Mr1.1与α9α10 nAChR不同位点的结合模式
为了进一步研究Mr1.1的作用机制和构-效关系,研究者们分别采用多肽的丙氨酸扫描、相似氨基酸残基替换、基于人工智能技术的复合物模型构建与分子动力学模拟方法,研究了Mr1.1中影响其与受体(α9α10 nAChR)结合的关键性残基及其作用机制(图2),并将这些数据与Vc1.1的已有研究进展进行对比(Chu and Yu* et al, ACS Chem Neurosci. 2019, 10(10):4328-4336),从而指导Mr1.1的结构修饰与改造。研究数据表明:Mr1.1能够与α9α10 nAChR形成多个氢键、盐桥和范德华力等相互作用。而后基于CADD模型和数据分析,研究者们设计合成了多个Mr1.1的突变体,希望通过增强分子间作用力来增强Mr1.1的活性。经活性测试,终于获得了多个受体抑制活性显著增强的Mr1.1类似物。其中Mr1.1[S4Dap]对人α9α10 nAChR靶点抑制活性IC50为4nM,是曾进入临床二期研究的Vc1.1活性的250倍(Yu* et al. J Med Chem. 2018,61(10):4628-4634)。最后,通过坐骨神经慢性压迫损伤模型,研究者们评估了Mr1.1[S4Dap]的体内镇痛活性:Mr1.1[S4Dap]能够在0.5 μg/kg的给药剂量下就表现出明显的镇痛活性,而25 μg/kg的给药剂量组甚至显示出强于阳性药加巴喷丁的体内镇痛活性(图3)。
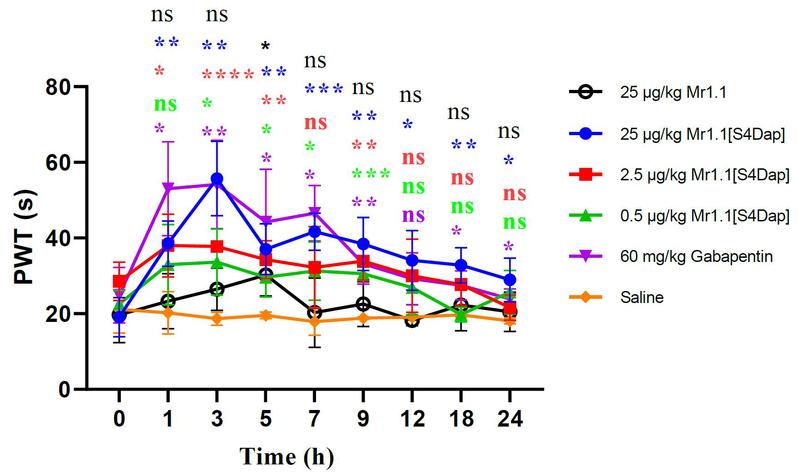
图3:芋螺毒素Mr1.1及相关类似在坐骨神经痛动物模型上镇痛活性
以上研究为芋螺毒素的作用机制研究以及合理的结构修饰提供了指导和建议,也为后续镇痛药的开发提供了极具开发潜力的先导分子。中国海洋大学医药学院为本论文第一通讯单位,医药学院博士研究生梁家珍和卧龙岗大学Han-Shen Tae为该文章第一作者,江涛教授、陈棽博士、李晓、赵紫彤和张景会为本论文的共同作者,卧龙岗大学David Adams教授和于日磊教授为该文章的通讯作者。该研究由国家自然科学基金面上与优秀青年基金等多个项目资助。
【关闭】





