近日,中国海洋大学医药学院海洋药物教育部重点实验室于日磊教授课题组,基于前期建立的芋螺多肽智能发现技术(Zhang et al, Acta Pharmaceutica Sinica B, 2025),率先发现了作用α9α10 nAChR的芋螺多肽孔道阻滞剂。相关研究成果以“Discovery and optimization of a monodisulfide conotoxin defines a potent and selective α9α10 nAChR blocker with analgesic efficacy”为题,在国际药物化学领域顶级期刊Journal of Medicinal Chemistry在线发表(DOI: 10.1021/acs.jmedchem.5c03296)。
海洋天然产物因其独特的化学结构和强靶向性,一直是创新药物研发的重要源泉。芋螺毒素作为海洋软体动物芋螺分泌的一类富含二硫键的生物活性肽,能够精准靶向细胞膜上的离子通道和受体,具有极高的药用价值。其中,α9α10乙酰胆碱受体(α9α10 nAChR)已被证实是治疗神经病理性疼痛的关键靶点。然而,目前已报道的作用于该靶点的芋螺毒素大多存在亚型选择性差等缺点,严重制约了其临床转化。
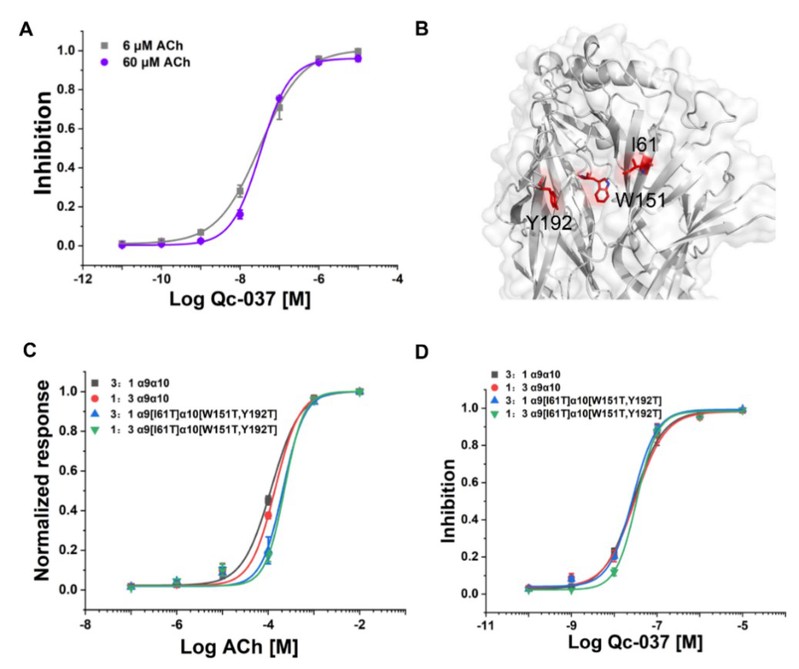
图1. Qc-037与α9α10 nAChR相互作用的机制
针对这一科学难题,研究团队独辟蹊径,基于AI识别技术锁定一个此前未被充分研究的Z超家族芋螺毒素Qc-037。该毒素结构特殊,仅含一对二硫键(单二硫键),与传统芋螺毒素的二到四对二硫键骨架截然不同。通过电生理验证和人工智能结构预测,团队首次发现Qc-037并非像经典的芋螺毒素(如Vc1.1)那样占据乙酰胆碱结合的正构位点,而是作为一种孔道阻断剂,直接物理性地堵塞该离子通道型受体。实验证明,该抑制作用不依赖于乙酰胆碱浓度,也不受受体亚基化学计量比((α9)3(α10)2与(α9)2(α10)3)的影响,这一独特机制有望规避因体内受体亚基化学计量比例不明而导致的临床失效风险(图1)。
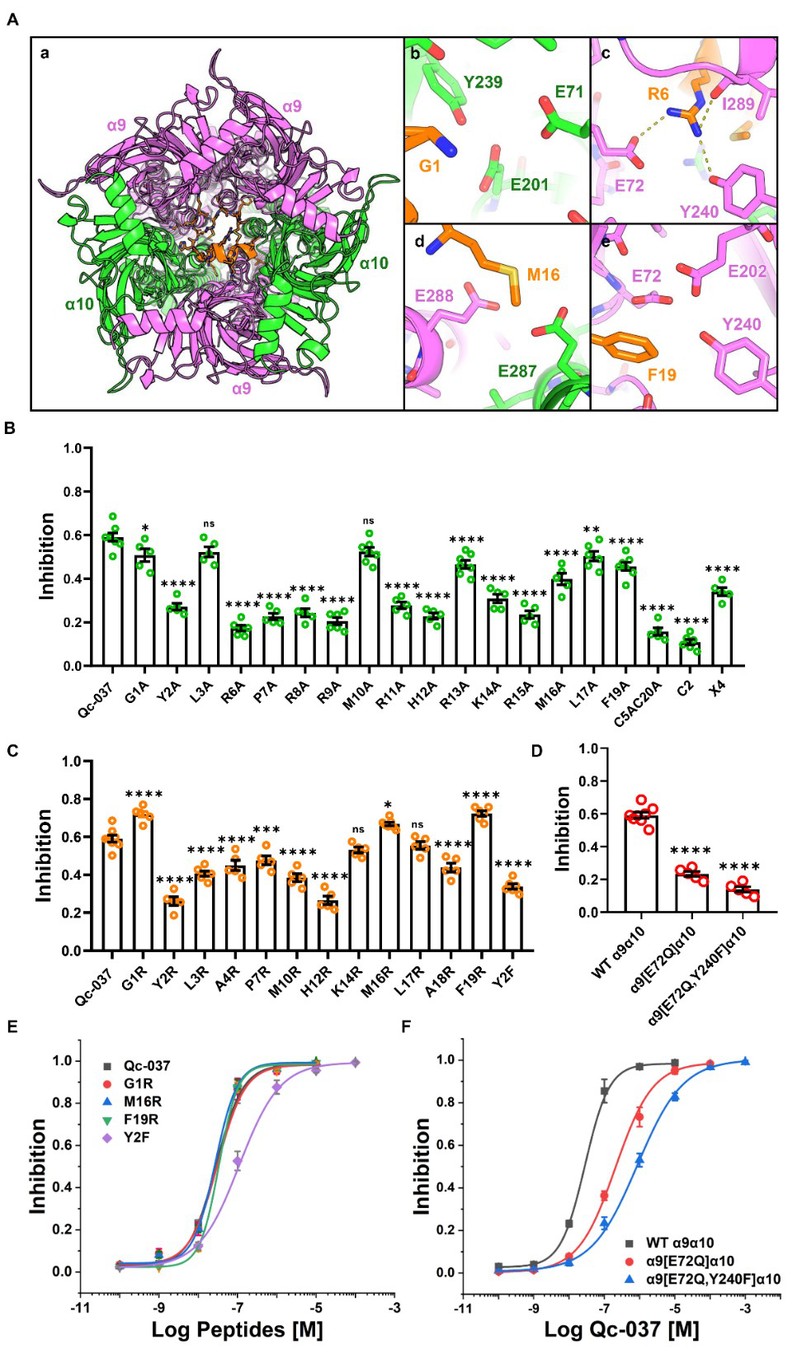
图2. Qc-037与α9α10 nAChR孔道的结合模式及结构-活性关系
基于结构的理性设计显著提升药效:在阐明作用机制的基础上,团队深入开展了构效关系研究,对Qc-037的每一个氨基酸进行了系统的丙氨酸和精氨酸扫描。通过解析多肽与受体的结合模式,以结构为指导研究人员成功设计出高选择性突变体[M16R]和[F19R]。这两个改造后的多肽不仅保持对α9α10 nAChR的抑制活性,更实现了选择性突破,有效降低了脱靶副作用风险(图2)。此外,在奥沙利铂诱导的冷异常性疼痛(化疗诱导神经病理性疼痛模型)大鼠模型中,[M16R]和[F19R]表现出显著的镇痛效果,且在部分测试中作用持续时间长于阳性对照肽RgIA4(图3)。
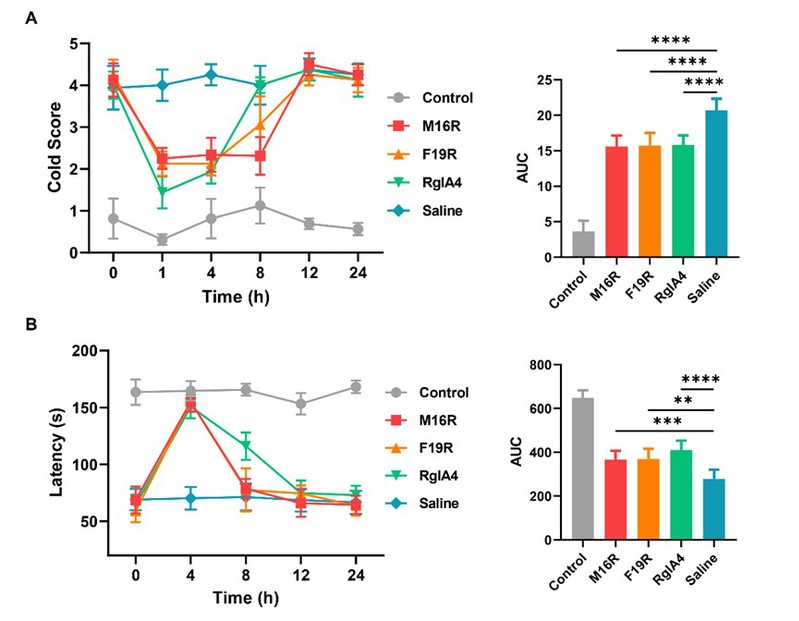
图3. [M16R]和[F19R]对奥沙利铂引起的冷觉异常的镇痛作用
研究团队通过电生理学、多肽药物化学和动物行为学的紧密结合,不仅发现了全新作用机制的芋螺毒素先导分子,更为重要的是,展示了如何利用结构信息对一个天然产物进行精准优化,从而攻克活性和亚型选择性无法兼顾的难题。这种“孔道阻断”的机制也为开发新型镇痛药物提供了区别于传统竞争性拮抗剂的替代策略。
中国海洋大学医药学院为该文章第一通讯单位,于日磊教授为该文章的通讯作者,医药学院博士研究生尹正基和医药学院毕业生宋雪为该文章第一作者,江涛教授、周缨、秦臻博士、袁璞、王纯和王晨宇为该文章的共同作者。该研究得到了国家自然科学基金、中央高校基础研究经费以及青岛市科技示范工程等多个项目的联合资助。
【关闭】





